
Often, when trying to find out what’s going on within our favourite green friends, we scientists simply grab a plants, grind it up, and perform a range of molecular or biochemical tests. But if we’re completely honest with ourselves, we’ll acknowledge that not all plant parts are created equal.
Leaves get all into photosynthesis, and roots have specialised bits that help them snuggle into the earth in search of nutrients. Within these ‘leafy’ or ‘rooty’ organs, there are also further subgroups: specialised types of tissue or cell. Plant leaves for example, have epidermal cells for protection, guard cells that open and close stomatal pores to let air in and out, palisade cells packed with chloroplasts for maximal light absorbtion, and xylem and phloem that transport water and nutrients (to name just a few).
In the end, what makes we complex organisms ‘special’, is the fact that we have developed 1000s of different cell types that all have specialized form and function, but work together to form a whole. But this means, that if we want to understand how plants really work, we need to know how all the cells work, and particularly how they all work differently from eachother. And here, the grab and blend method of ‘bucket biology’, simply isn’t going to work.
Enter single celled biology.
In which, as the name suggests, you take a single cell, and perform analysis of just that cell.
(And then you do the same thing with thousands of other cells, to find out how different they are from eachother).
Single celled biology, as a field, is still in its infancy, because we need to have technology (machines) that are sensitive enough to measure stuff at tiny scales. (Usually, half of the work of being a biologist is just collecting enough material to make the measurements that you need!) But in recent years, single celled transcriptomics has become possible.
Transcriptomics, aka RNA sequencing, is itself already pretty amazing- it lets us see exactly how much of each type of RNA there is in an organism. This in turn, gives us clues about of which genes are being turned on and which are staying turned off. Now, with the current RNA sequencing technology, coupled with a few nifty preparation tricks, it’s possible to understand the expression of genes on a cell-by-cell basis.
One of these methods, named ‘Drop-Seq’, involves co-capturing individual cells and special microparticles in tiny oily droplets. This process involves pushing the microparticles down a highway, until the intersect with the cells. Further down the road, the particle/cell mixture meets up with some oil, which forces everything into contained drops.
Something like this:
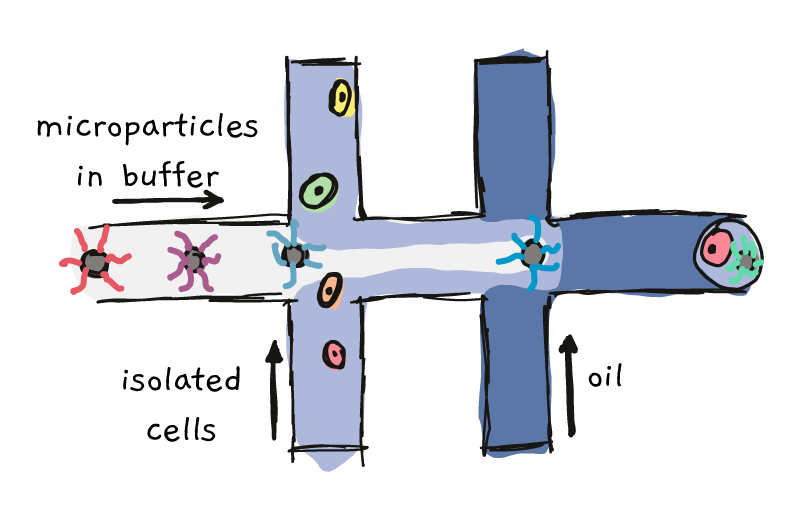
With the right settings, each oily drop contains one single cell and one single microparticles. Each of the microparticles is covered in marker sequences, called barcodes. All the barcodes on one particle are the same, but from particle to particle, the barcodes are different. Once the particle and cell get together in their droplet, lysing (breaking) of the cell, followed by a few extra steps, allows the RNA to become labeled with the barcode.
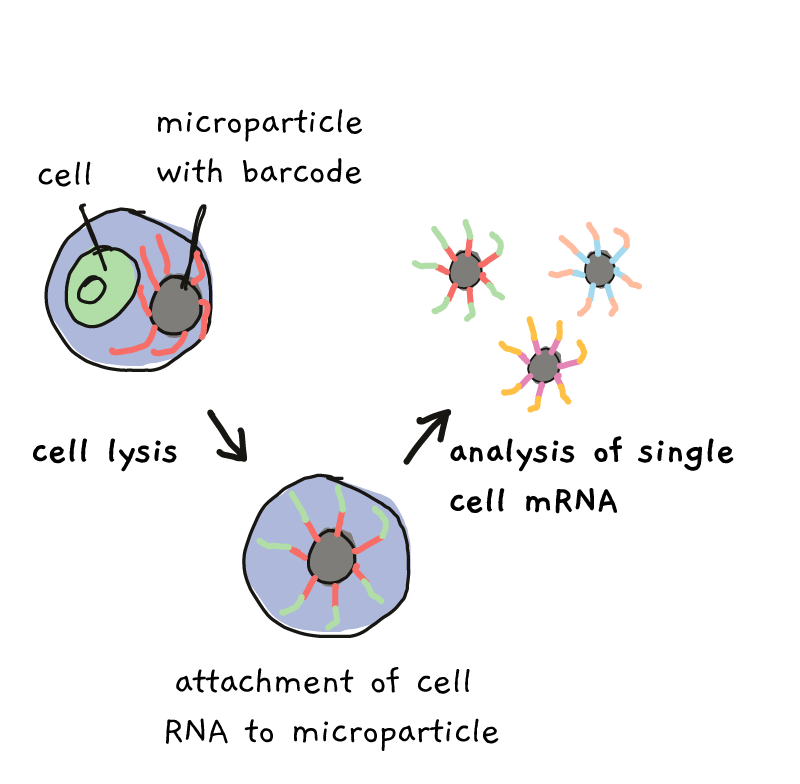
By the end of the process, all of the RNA from one cell, in one droplet, has one barcode, while RNA from a different cell/droplet has a different barcode. Everything can be mixed together and sequenced, but at the end, those barcodes tell you who came from which cell.
Despite how amazing this technique is, up until now there hasn’t been a lot of single celled transcriptomics in the plant field. That’s because in addition to plant cells being generally larger and more irregularly sized than the mammalian cells that Dropseq was designed for, they also have cell walls.
So in order to do Dropseq, you first have to convince plants to drop their cell walls.
In a recent publication, Shulse and collegues wanted to look at transcript expression in different root cell types. So they first grew a whole lot of roots and chopped them up. Then they added pectolyase and cellulysin to the mix, enzymes which both work to destroy cell walls. Once they had these little naked cells (known as protoplasts), they proceeded with the Drop-seq-ing: individually barcoding more than 12,000 different root cells that all got sent off for RNA sequencing.
So what could they do with all that data? Well firstly, they looked at the 12,000 cells and tried to work out which ones behaved similarly. They reasoned that cells that express similar amounts of the same mRNA are likely acting in the same way (i.e., they’re the same cell type), while other cells with different mRNA expression are probably different cell types. In the end, they came up with 17 clusters of cells. By looking at the features of these types, they realised that their 17 groups represented most of the known root cell types, such as epidermal hair cells, lateral root cap cells, and so on. But they also noticed that many of these known root cell types were represented by more than one of the 17 groups. Suggesting that within some of the previously defined root cell types, there actually exists two or more ‘subtypes’ of cell, which might have slightly different functions from eachother.

Another strange finding was that many of the genes previously used as markers for certain cell types, were not very reliable. A ‘marker’ gene should be something that is expressed in one cell type but not others, thus ‘marking’ that certain type. But many of the known markers were either weakly expressed in the tissue they marked, or- worse-, were actually expressed in multiple tissue types. So the authors of the study suggested new markers that could be used for future work.
In addition to looking at different cell types, Shulse and collegues looked at the effect of growing plants with and without sucrose in the media. Sucrose has been shown to change aspects of plant growth, and yet scientists commonly either include or omit sucrose based entirely on personal preference. Although sucrose addition didn’t change the type of cell populations found in roots, the proportion of cells belonging to each of the 17 cell types did change, and in general, roots grown on sucrose looked more mature. Furthermore, nearly 2000 genes were differentially regulated depending on whether the plants had a sweet snack or not.
Overall, this work largely highlights just how far we’ve come with recent technology.. but also shows us just how much we still don’t know about the wonderful world of plants.
References
This journal club was based on:
High-Throughput Single-Cell Transcriptome Profiling of Plant Cell Types
Christine N. Shulse, Benjamin J. Cole, Doina Ciobanu, Junyan Lin, Yuko Yoshinaga, Mona Gouran, Gina M. Turco, Yiwen Zhu, Ronan C. O’Malley, Siobhan M. Brady, and Diane E. Dicke
The DropSeq paper, which looked at mice retinas!!
Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets
E.Z. Macosko, A. Basu, R. Satija, J. Nemesh, K. Shekhar, M.Goldman, I. Tirosh, A.R. Bialas, N. Kamitaki, E.M. Martersteck, et al. Cell, 161 (2015), pp. 1202-1214
Check out a discussion about “The potential of single-cell profiling in plants”:
https://genomebiology.biomedcentral.com/articles/10.1186/s13059-016-0931-2